Cada producto necesita un mercado. También necesita acceso al mercado. Para los productos farmacéuticos, eso significa HTA, reembolso, fijación de precios, posicionamiento y otros elementos que pueden marcar la diferencia entre un lanzamiento espectacular y un costoso lanzamiento fallido. En nuestras tendencias farmacéuticas para 2024, analizamos algunos desafíos críticos de acceso al mercado en territorios clave para la industria y cómo las empresas pueden prepararse mejor para ellos durante el próximo año.
La mala noticia es que el acceso al mercado no es cada vez más fácil. Esto refleja varias tendencias más amplias que están dando forma a la industria farmacéutica, como la presión incesante sobre las finanzas y los recursos del sistema de salud, exacerbada por el COVID-19, el envejecimiento de la población y las enfermedades relacionadas con la edad; conveniencia política en la financiación y la toma de decisiones en materia de salud; terapias más complejas y costosas; el continuo aumento del poder de los pagadores; y beneficios digitales inesperados, como un rápido intercambio de información y análisis más sofisticados.
Para las empresas realmente sintonizadas con lo que se avecina y cómo abordarlo tanto a nivel global como local, estos no son sólo desafíos sino diferenciadores potencialmente cruciales en un mercado ferozmente competitivo. Sin un acceso óptimo al mercado farmacéutico, la excelencia en el lanzamiento solo puede ser una ambición con una relevancia limitada en el mundo real para las empresas, los accionistas, los sistemas de salud, los profesionales de la salud o los pacientes.
Tendencias farmacéuticas para 2024
Tendencia #1. Revisión farmacéutica de la UE: nuevos desafíos para los equipos de acceso al mercado
Si tuviera un medicamento nuevo y emocionante, aprobado centralmente para su uso en los 27 estados miembros de la UE, ¿no le gustaría lanzarlo en tantos mercados como sea posible y lo más rápido posible, llegando al máximo número de pacientes? Teóricamente sí; en la práctica, no necesariamente.
El acceso de los pacientes y el acceso a los mercados a veces tienen que separarse. Por supuesto, el acceso de los pacientes es fundamental para el acceso al mercado de un nuevo lanzamiento. Pero el acceso al mercado también consiste en optimizar el potencial comercial . Eso significa lanzarlo en el momento adecuado y con el precio, el reembolso y la propuesta de valor adecuados. La flexibilidad para hacerlo, en un mercado de la UE nominalmente armonizado, va en contra de las nuevas propuestas para cuadrar el mercado y el acceso de los pacientes en toda Europa.
En realidad, los precios, el reembolso y los procedimientos de ETS son tendencias de la industria farmacéutica sujetas a grandes variaciones, tanto a nivel mundial como dentro de la UE. La financiación disponible para nuevos medicamentos depende en parte de las prioridades sanitarias y de las economías locales. Las capacidades de ETS pueden estar más evolucionadas en algunos países que en otros, con diferentes énfasis en sus criterios de evaluación (impacto presupuestario, costo-beneficio, etc.). Los sistemas de fijación de precios y reembolso tienen sus propias metodologías, requisitos y plazos.
Esto complica significativamente los lanzamientos paneuropeos y su secuenciación. La programación del lanzamiento también debe tener en cuenta el potencial de referenciación de precios de un Estado miembro a otro y de importaciones paralelas desde mercados de precios más bajos a mercados más altos. En consecuencia, es posible que una empresa desee priorizar los lanzamientos en estados miembros con precios y reembolsos más permisivos.
Incluso puede optar por retrasar o renunciar al lanzamiento en algunos mercados, si los precios y reembolsos disponibles no sólo son antieconómicos sino que pueden socavar la entrada al mercado en otros estados miembros. Esto ha sucedido varias veces recientemente en Alemania (tradicionalmente un producto de comercialización), y particularmente con los nuevos medicamentos contra el cáncer (ver más abajo).
Refleja en parte los cambios legislativos en Alemania que han puesto freno a la libertad de fijación de precios y han vinculado el reembolso de los medicamentos de manera más estricta al valor agregado. La tendencia también es sintomática de un sistema de HTA que ha rechazado la supervivencia libre de progresión como un criterio de valoración sustituto digno de calificaciones de beneficios adicionales en oncología.
La revisión farmacéutica de la UE
Este es el contexto de quizás la más polémica de las propuestas de la amplia revisión de la legislación farmacéutica de la UE por parte de la Comisión Europea, publicada el 26 de abril de 2023 y ahora ante el Parlamento Europeo. La Comisión quiere reducir la protección de datos reglamentaria (PDR) básica para nuevos medicamentos de dos a seis años, más dos años más de exclusividad en el mercado.
El PDR perdido se recuperaría si los medicamentos estuvieran disponibles para su suministro continuo en todos los estados miembros dentro de los dos años posteriores a su aprobación. El período de protección también podría ampliarse si los medicamentos cumplieran otros criterios, como abordar necesidades médicas no cubiertas o realizar ensayos clínicos comparativos (ambos restableciendo el PDR de seis meses).
Esta postura más coercitiva refleja un objetivo central de la revisión farmacéutica de la Comisión: garantizar que todos los pacientes tengan acceso oportuno y equitativo a medicamentos seguros, eficaces y asequibles. Sin embargo, el enfoque del palo y la zanahoria plantea inmediatamente la cuestión de si los lanzamientos paneuropeos en un plazo de dos años son siquiera alcanzables, dado que dependen en parte de la discreción y los recursos de los Estados miembros individuales.
En algunos de estos países, los procedimientos de fijación de precios y reembolso ciertamente pueden ser lentos. El último indicador Patient WAIT de la Federación Europea de Industrias y Asociaciones Farmacéuticas (EFPIA) encontró que, en promedio, los nuevos medicamentos llegaban a los pacientes dentro de los 128 días posteriores a la aprobación centralizada en Alemania, 436 días en Italia, 629 en España, 918 en Rumania y 1 , 351 en Malta . El tiempo medio de disponibilidad en la UE en 2018-201 fue de 517 días.
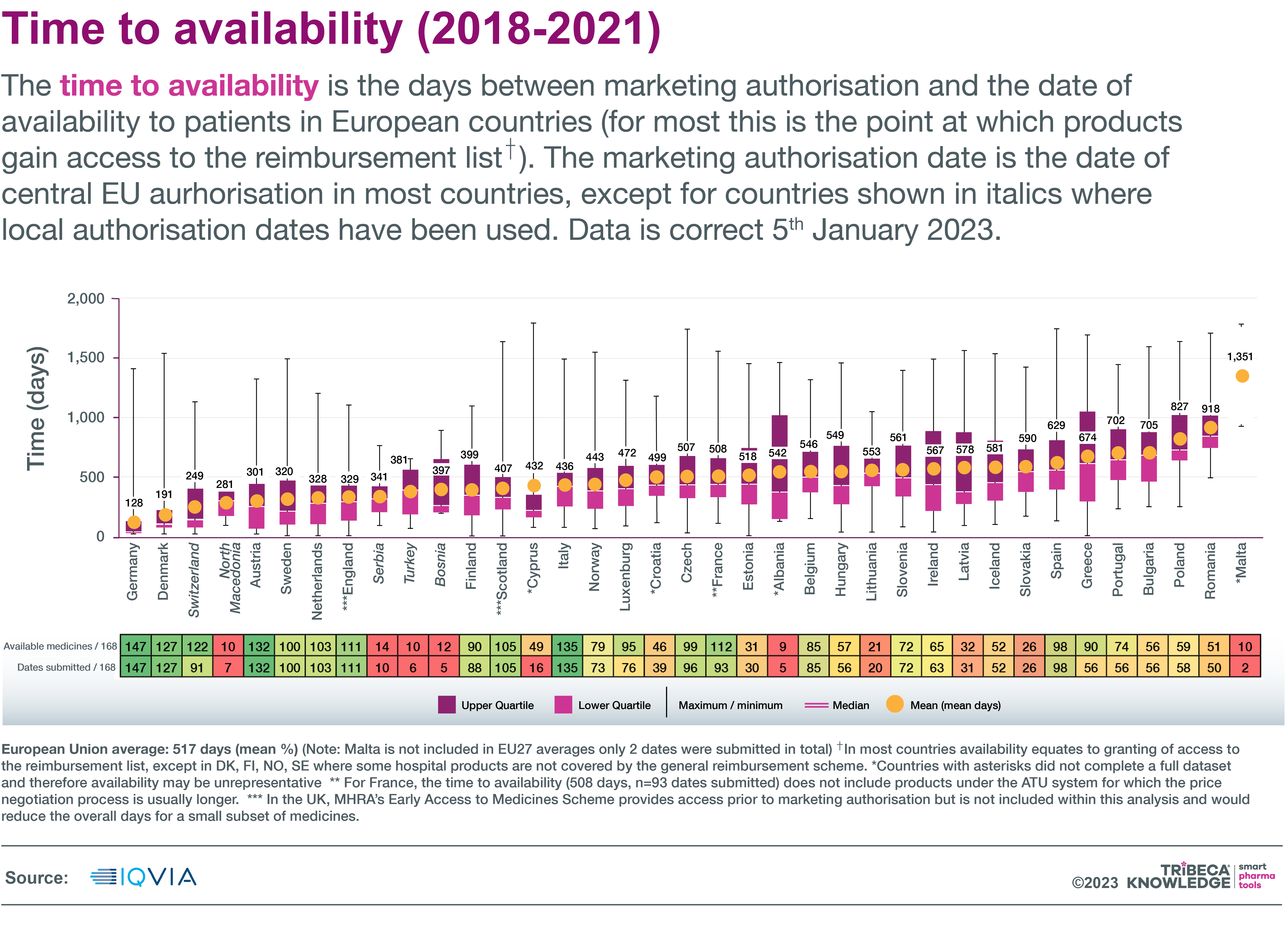
Fuente: IQVIA
¿Por qué ocurren retrasos?
Datos más recientes del Portal Europeo de Obstáculos de Acceso liderado por la industria sugieren que sólo alrededor de una cuarta parte del retraso en la disponibilidad de los pacientes se debe a retrasos en las presentaciones de las empresas. El resto se debe a retrasos en los procesos nacionales de fijación de precios y reembolsos, afirma EFPIA.
Incluso las presentaciones tardías de acceso a los mercados se deben a diferentes razones : principalmente procesos de evaluación de valor y requisitos de evidencia en Europa occidental, mientras que en Europa del este los retrasos se deben más a limitaciones del sistema de salud y su impacto en la toma de decisiones corporativas. » Esto demuestra que no existe una solución única para solucionar los problemas de acceso, y que se necesitan soluciones regionales o específicas de cada país «, afirma EFPIA.
También se teme que la ventana de lanzamiento de dos años pueda resultar en problemas de suministro de medicamentos o que, con el tiempo corriendo, las empresas puedan verse presionadas a aceptar precios desfavorables o reembolsos en algunos mercados como condición de entrada. Además, EFPIA estima que un PDR de referencia reducido reduciría el incentivo para que las empresas inviertan en medicamentos innovadores en Europa en un 55% durante los próximos 15 años. Estamos en un momento en el que se prevé que la participación de la región en la inversión mundial en I+D caiga del 32% al 21% para 2040.
Disposiciones más alentadoras
Hay disposiciones más favorables a la innovación para la industria biofarmacéutica en el paquete farmacéutico, como la reducción del tiempo necesario para evaluar y aprobar nuevos medicamentos a nivel central en la UE. También hay planes para crear «zonas de pruebas» regulatorias, con marcos adaptativos que incluyan la recopilación de datos del mundo real, para acelerar la aprobación de terapias novedosas.
Sin embargo, los plazos más cortos del PDR no son la única preocupación de la industria. EFPIA ha cuestionado, por ejemplo, el alto listón para satisfacer necesidades médicas insatisfechas o altas para obtener RDP adicional o exclusividad en el mercado huérfano; y requisitos más estrictos para el desarrollo oportuno de medicamentos huérfanos.
Tal como están las cosas, las revisiones pueden estar lejos de su implementación. El paquete farmacéutico ya tuvo un comienzo tardío en 2023, mientras que las posibilidades de que el Parlamento Europeo y el Consejo completen sus discusiones antes de las elecciones parlamentarias de 2024 parecen escasas . La cuestión se complica aún más por las diferencias emergentes entre los Estados miembros sobre cuestiones como el equilibrio adecuado entre el acceso de los pacientes a los medicamentos y los incentivos para la innovación.
También es posible que se produzcan cambios importantes en el futuro. Un informe reciente sobre las propuestas de la Comisión elaborado por la ponente parlamentaria Pernille Weiss recomendaba, por ejemplo, un período de referencia del PDR más largo (de ocho a nueve años), junto con ajustes al requisito de lanzamiento oportuno y cambios en la definición de necesidad médica no cubierta que da derecho a una extensión ampliada. RDP.
Cualquiera que sea el resultado de estas deliberaciones, podrían marcar la pauta para las tendencias de acceso al mercado en 2024 y hacer de la Unión Europea un campo de batalla clave para lanzar estrategias de excelencia que satisfagan los intereses comerciales y de los pacientes sin socavar ninguno de los dos.
Tendencia #2. Las presiones sobre los precios se intensifican: ¿Todo lo que el IRA puede hacer, Alemania y Japón pueden hacerlo mejor?
Las disposiciones sobre fijación de precios de los medicamentos contenidas en la Ley de Reducción de la Inflación del año pasado marcaron un alejamiento significativo del modelo de libre fijación de precios que convirtió a Estados Unidos en un importante mercado de lanzamiento de nuevos medicamentos. El IRA dio laLos Centros de Servicios de Medicare y Medicaid tienen autoridad para negociar Precios Justos Máximos (MFP) para los medicamentos de alto costo utilizados en el esquema de Medicare. También incluía sanciones de reembolso para las empresas que elevaran los precios de los medicamentos de Medicare por encima de la inflación.
Entre las tendencias actuales más exigentes en la industria farmacéutica, las multinacionales acostumbradas a lidiar con controles de precios complejos y variables en mercados clave como Europa o Japón pueden haber sentido que la vista desde el otro lado del Atlántico o el Pacífico no parecía tan mala después de todo. . El entorno de precios en Europa y Japón se está volviendo cada vez más difícil.
Las impresoras multifunción entran en acción
El primer lote de 10 medicamentos de la Parte D sujetos a MFP, incluidos éxitos de taquilla como Eliquis (apixaban) de Bristol-Myers Squibb y Jardiance (empagliflozina) de Eli Lilly, se anunció en agosto de 2023 . Los MFP negociados entrarán en vigor el 1 de enero de 2026. Los descuentos mínimos aplicados a los medicamentos elegibles para los MFP oscilarán entre el 25% y el 60%, dependiendo de cuánto tiempo lleve un medicamento en el mercado. Las sanciones por aumentos de precios que reducen la inflación ya están al alcance de la mano: en junio de 2023 se anunciaron reembolsos de segunda ronda para 43 medicamentos.
El impacto más amplio de estos controles en el acceso al mercado estadounidense, las estrategias comerciales y el desarrollo de fármacos sigue siendo incierto. Dependerá en parte de factores como el grado de exposición de un activo al mercado de Medicare y cualquier posible derrame o compensación con los segmentos del mercado comercial.
Incluso podría suceder algo peor para la industria farmacéutica . Las propuestas de presupuesto federal del presidente Biden para el año fiscal 2024 incluyen planes para duplicar la cantidad de medicamentos sujetos a negociación de precios de Medicare, reducir e igualar los períodos de gracia tanto para los productos biológicos como para los medicamentos de molécula pequeña, y extender las sanciones por aumentos de precios superiores a la inflación a los productos comerciales. sector.
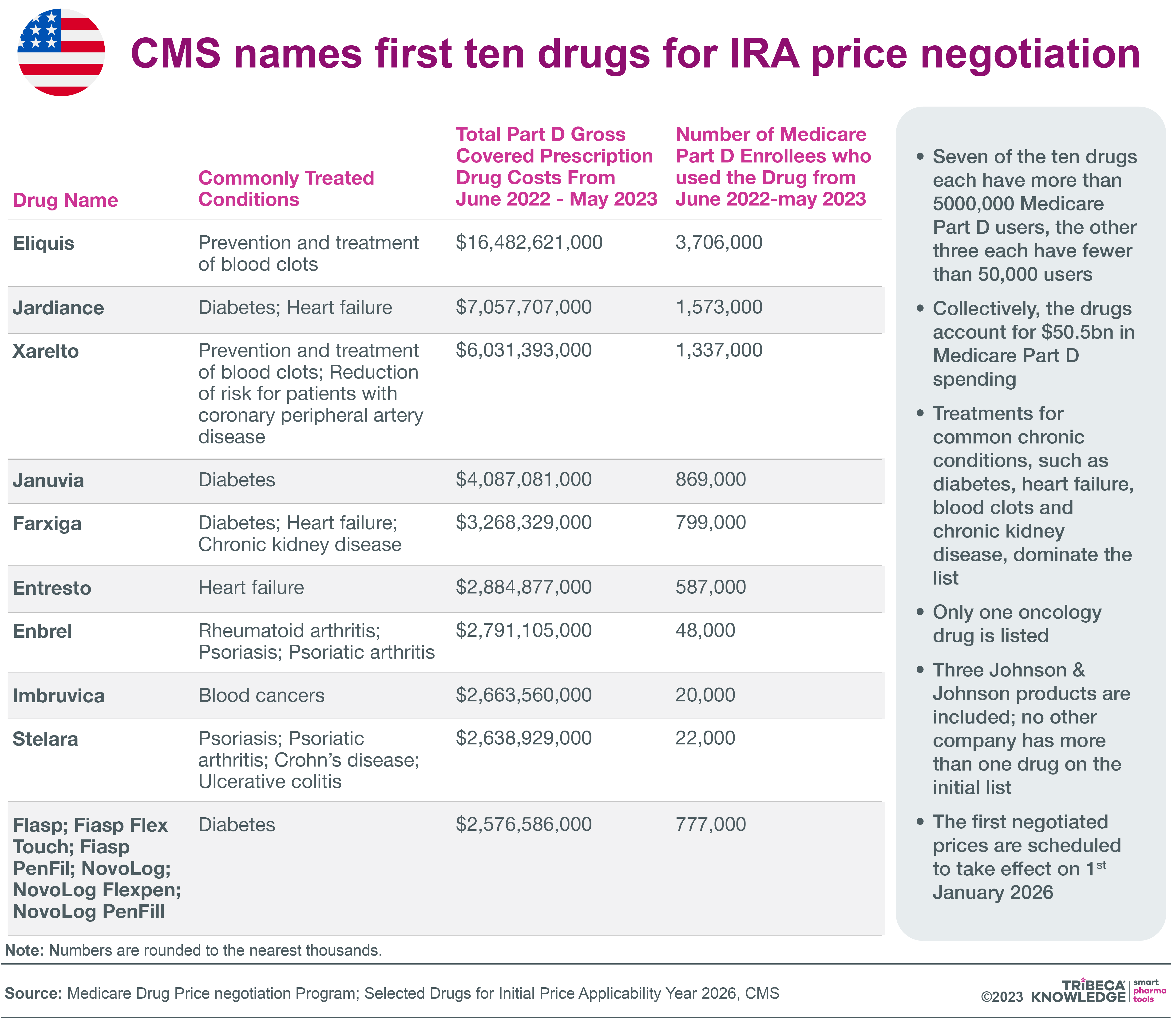
Fuente: Neil Grubert
Clima frío en Alemania
Más allá, un acontecimiento reciente con implicaciones para los precios de los medicamentos en toda la UE son las propuestas de la Comisión para una reforma farmacéutica de amplio alcance (ver arriba). Estos no abordan directamente el precio de los medicamentos; esto sigue siendo, junto con las políticas de reembolso, una responsabilidad nacional. Sin embargo, las propuestas corren el riesgo de socavar las estrategias de lanzamiento impulsadas por las discrepancias de precios en toda la región y sus efectos dominó sobre los precios de referencia o las importaciones paralelas.
Por ejemplo, la Comisión quiere reducir los períodos regulatorios de protección de datos y de exclusividad en el mercado para nuevos medicamentos, a menos que se lancen en todos los estados miembros dentro de los dos años posteriores a su aprobación . Esto debería mejorar la equidad de acceso para los pacientes, pero también podría diluir las estrategias de lanzamiento preferenciales orientadas a mercados tradicionalmente de precios más altos, como Alemania. Dicho esto, incluso el mercado alemán está empezando a perder su atractivo.
La Ley de Estabilización Financiera del Sistema Obligatorio de Seguro de Enfermedad ( GKV-FinStG ), que entró en vigor en noviembre de 2022, incluyemedidas como :
- Reducir de 12 a seis meses el período de libre precio de los medicamentos de nuevo lanzamiento
- Reducir a 30 millones de euros el umbral de facturación anual por encima del cual los medicamentos huérfanos están sujetos a evaluaciones de beneficios completos.
- Un aumento de un año al 12% de las ventas en el descuento obligatorio a las aseguradoras de salud obligatorias
- Un reembolso obligatorio del 20% en nuevas terapias combinadas
- ‘Barreras’ de fijación de precios más estrictas para medicamentos sin beneficios añadidos significativos: por ejemplo, el precio de reembolso de un medicamento sin beneficios añadidos debe ser al menos un 10 por ciento inferior al del comparador no genérico más económico.
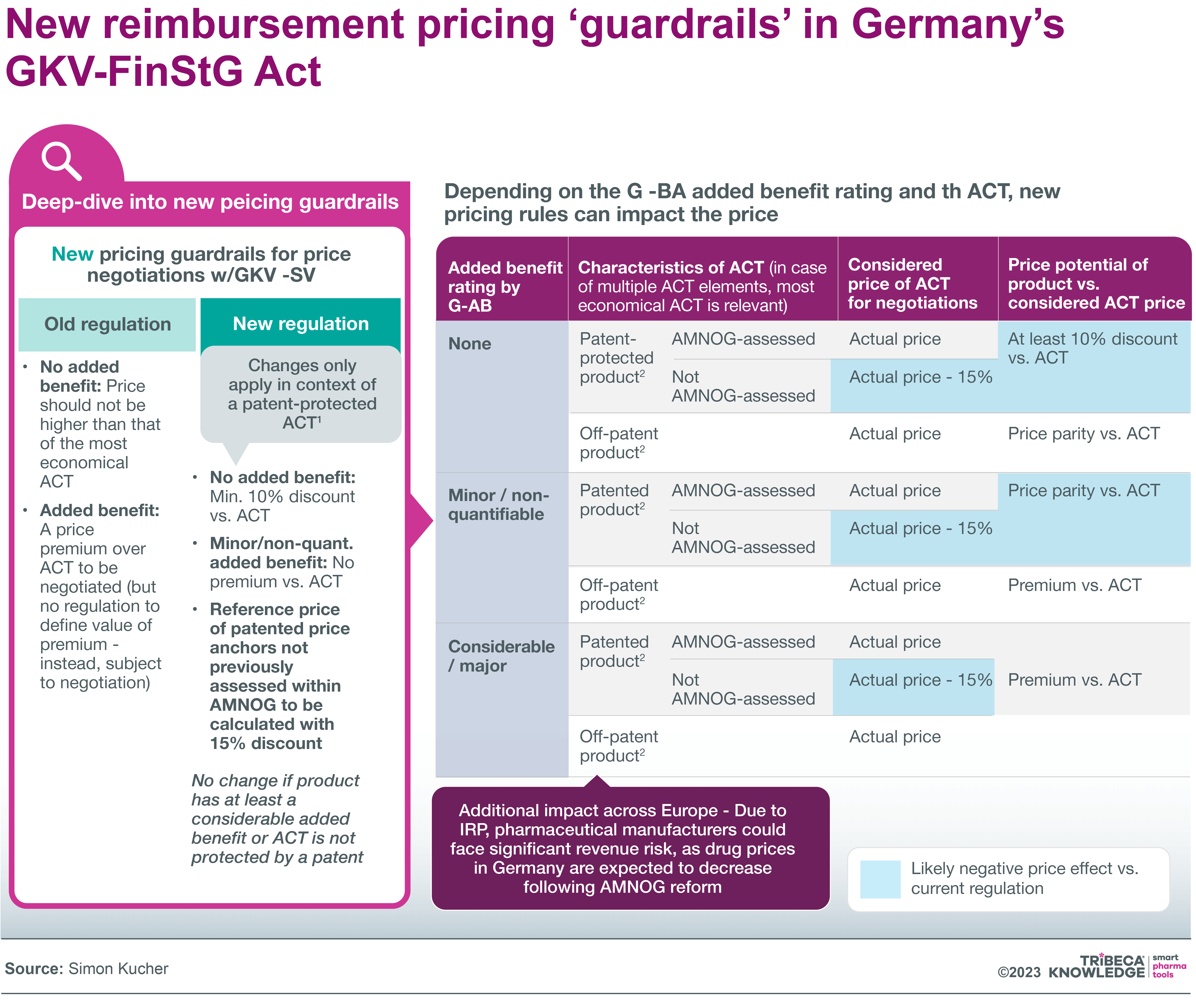 Fuente: Simon Kucher
Fuente: Simon Kucher
Se ha culpado a las nuevas disposiciones de precios y reembolsos por una serie de decisiones de no lanzar o retirar medicamentos del mercado. Estos incluyen el anticancerígeno Opdualag (nivolumab + relatlimab) de Bristol-Myers Squibb ; Spevigo (spesolimab) de Boehringer Ingelheim para los brotes de psoriasis; y Tabrecta (capmatinib), el medicamento contra el cáncer de pulmón de Novartis . Una encuesta reciente realizada por la asociación de investigación VFA encontró que 21 de 48 empresas miembro habían decidido retrasar o evitar el lanzamiento de nuevos medicamentos/indicaciones en Alemania, o estaban considerando hacerlo, como resultado directo de las medidas de GKV-FinStG .
Japón duplica sus recortes de precios
Japón es otro mercado importante en el que la industria farmacéutica está sintiendo la presión de los precios. En 2021 se implementaron revisiones adicionales de precios fuera de año para los productos farmacéuticos , además de las revisiones bienales habituales de Japón. Reaparecieron en 2023. Las últimas revisiones fuera de año entraron en vigor el 1 de abril de 2023 y se aplicaron a los medicamentos con discrepancias. entre los precios de mercado y los precios del Seguro Nacional de Salud que superan 0,625 veces la tasa de variación de precios promedio en 2022
Esto llevó a reducciones de precios en más de 2.000 medicamentos en abril , y los medicamentos protegidos por patentes más afectados sufrieron recortes de precios promedio del 8-9%, según GlobalData. También se produjeron algunos aumentos de precios, ya que se introdujeron medidas especiales para garantizar un suministro estable de determinados medicamentos. Esto significó sobreprecios para 143 medicamentos innovadores o no rentables que se beneficiaron de sobreprecios, mientras que otros 90 medicamentos mantuvieron sus precios bajo el programa regular de primas de mantenimiento de precios.
Por lo demás, 240 productos vieron aumentar sus precios en la revisión de abril, una media cercana al 29%. Sin embargo, a pesar de estos avances, los precios del 91% de los medicamentos sujetos a revisión de precios en Japón se mantuvieron sin cambios o se redujeron, señaló GlobalData. Advirtió además que era poco probable que las medidas especiales de fijación de precios para medicamentos no rentables continuaran durante el año fiscal 2024 , lo que “solo aumentaría las ya fuertes críticas de los representantes de la industria al gobierno sobre la frecuencia de los ajustes de precios”.
Un panel de expertos del Ministerio de Salud, Trabajo y Bienestar Social se hizo eco de esas preocupaciones en un borrador de informe a principios de este año . Dado que Japón ha perdido su ventaja como innovador global en los últimos años, se debe alentar a la industria a asumir riesgos , sugirió el panel. Eso requirió una nueva reflexión sobre aspectos críticos del sistema de fijación de precios de los medicamentos, como mantener los precios mientras los nuevos medicamentos estaban bajo patente, en lugar de simplemente desgastarlos con revisiones anuales de precios.
Tendencia n.º 3. Preparándose para EU HTAR: es el momento decisivo…
A diciembre de 2023, a las empresas farmacéuticas les queda poco más de un año antes de que se produzcan cambios significativos en el panorama europeo de las evaluaciones de tecnologías sanitarias. Esto hace que 2024 sea un año crítico de ajuste y pensamiento creativo para los equipos de acceso al mercado sensibles a las principales tendencias de la industria farmacéutica y los caprichos del acceso al mercado de la UE.
Todavía quedan preguntas sin respuesta sobre cuánto logrará el Reglamento (UE) 2021/2282 de la Unión Europea sobre evaluación de tecnologías sanitarias (HTAR) , en vigor desde enero de 2022, a la hora de armonizar facetas de la ETS en toda Europa. También es discutible si esa armonización beneficiará a las compañías farmacéuticas que buscan evaluaciones tecnológicas más fluidas que lleguen al nivel nacional.
Sin embargo, las empresas no perderán nada si se aseguran de estar lo más informadas y preparadas posible para absorber el impacto de la regulación. Como advierten los consultores Deloitte , existe “una necesidad urgente de que las empresas farmacéuticas y de tecnología médica reconsideren la forma en que históricamente se han organizado internamente y se han preparado para las presentaciones de ETS específicas de cada país”.
El nuevo reglamento establece un marco legal y organizativo para la cooperación entre los estados miembros en evaluaciones clínicas conjuntas (JCA) a nivel de la UE de tecnologías sanitarias nuevas o existentes . El proceso JCA comienza con nuevos medicamentos contra el cáncer y medicamentos de terapia avanzada en enero de 2025, ampliándose a medicamentos huérfanos a partir del 13 de enero de 2028. Todos los demás medicamentos aprobados centralmente a través de la EMA estarán sujetos a JCA a partir del 13 de enero de 2030.
Todo esto lo supervisará un Grupo de Coordinación centralizado con representantes de las autoridades nacionales de ETS y aportes de una red de partes interesadas que incluye pacientes, profesionales de la salud y asociaciones industriales, así como sociedades clínicas/científicas y organizaciones de consumidores. El Reglamento HTA también crea un marco para consultas científicas conjuntas, la identificación de tecnologías sanitarias innovadoras y la cooperación voluntaria más allá del alcance del Reglamento.
Llenando el marco
Los tres años transcurridos entre la adopción del Reglamento (UE) 2021/2282 y el primer lote de JCA se dedicaron al consorcio voluntario EUnetHTA21 ( Red Europea de Evaluación de Tecnologías Sanitarias ) , junto con los estados miembros y la Comisión Europea, a completar los requisitos legales del reglamento. y marco organizacional con procesos y metodologías para la armonización de la ETS . EU netHTA21 dejó de operar en septiembre de 2023 y será reemplazado por el Grupo de Coordinación HTA
Lo que el reglamento de ETS no permite es un procedimiento de ETS único y centralizado que pueda resolver las disparidades persistentes en las evaluaciones en toda la UE. Los JCA se limitan a evaluar la seguridad y eficacia clínica comparativa, aunque la elección de productos de comparación y criterios de valoración para la evaluación se pueden realizar a nivel de la UE. Las decisiones sobre la rentabilidad, el valor de los medicamentos o el impacto presupuestario, junto con las negociaciones asociadas sobre precios y reembolsos, siguen siendo responsabilidad de los estados miembros individuales.
Los JCA tampoco son jurídicamente vinculantes para los Estados miembros, aunque la participación de la industria en el procedimiento es obligatoria. Los miembros solo están obligados a prestar la debida consideración a las JCA al realizar procedimientos nacionales de ETS. Es comprensible que estas áreas grises hayan puesto nerviosa a la industria.
Las complicaciones incluyen diferentes concepciones nacionales sobre los estándares de atención utilizados como comparadores de la efectividad clínica, o desacuerdos sobre la validez de criterios de valoración sustitutos aplicados en los ensayos clínicos. Aunque se espera que la regulación mejore las evaluaciones en los estados miembros donde la capacidad y experiencia en ETS están subdesarrolladas (por ejemplo, en Europa del Este), se teme que pueda llevar a una mayor duplicación del trabajo o complicar aún más el panorama del acceso al mercado.
Planifique con anticipación pero manténgase ágil
El consejo de Deloitte a los equipos de acceso al mercado es «planificar con anticipación pero mantenerse ágil», con especial atención a la preparación previa al lanzamiento, nuevos modos de toma de decisiones estratégicas, colaboración interfuncional y la participación de equipos locales. Si bien la regulación HTA requerirá cambios de extremo a extremo en el desarrollo de activos, lo que afectará a todos los equipos multifuncionales, el nivel de interrupción variará de un país a otro, dependiendo de la madurez actual y la voluntad de adaptarse a los procedimientos locales de HTA, dice Deloitte. .
A efectos de planificación, divide a estos países en cinco arquetipos: países salvaguardistas (por ejemplo, Alemania, Dinamarca), ahorradores de tiempo (Francia, Italia, Bélgica), descubridores (España, Portugal) y backbenchers (Lituania, Estonia, Grecia), además de países observadores fuera del país. la UE que puede tener en cuenta las JCA publicadas en sus propias evaluaciones. En los países Timesaver y Discoverer, debería haber más potencial para acelerar los procesos de HTA gracias a la regulación de HTA, incluso si esto implica una mayor interrupción para las presentaciones de HTA locales, señala Deloitte.
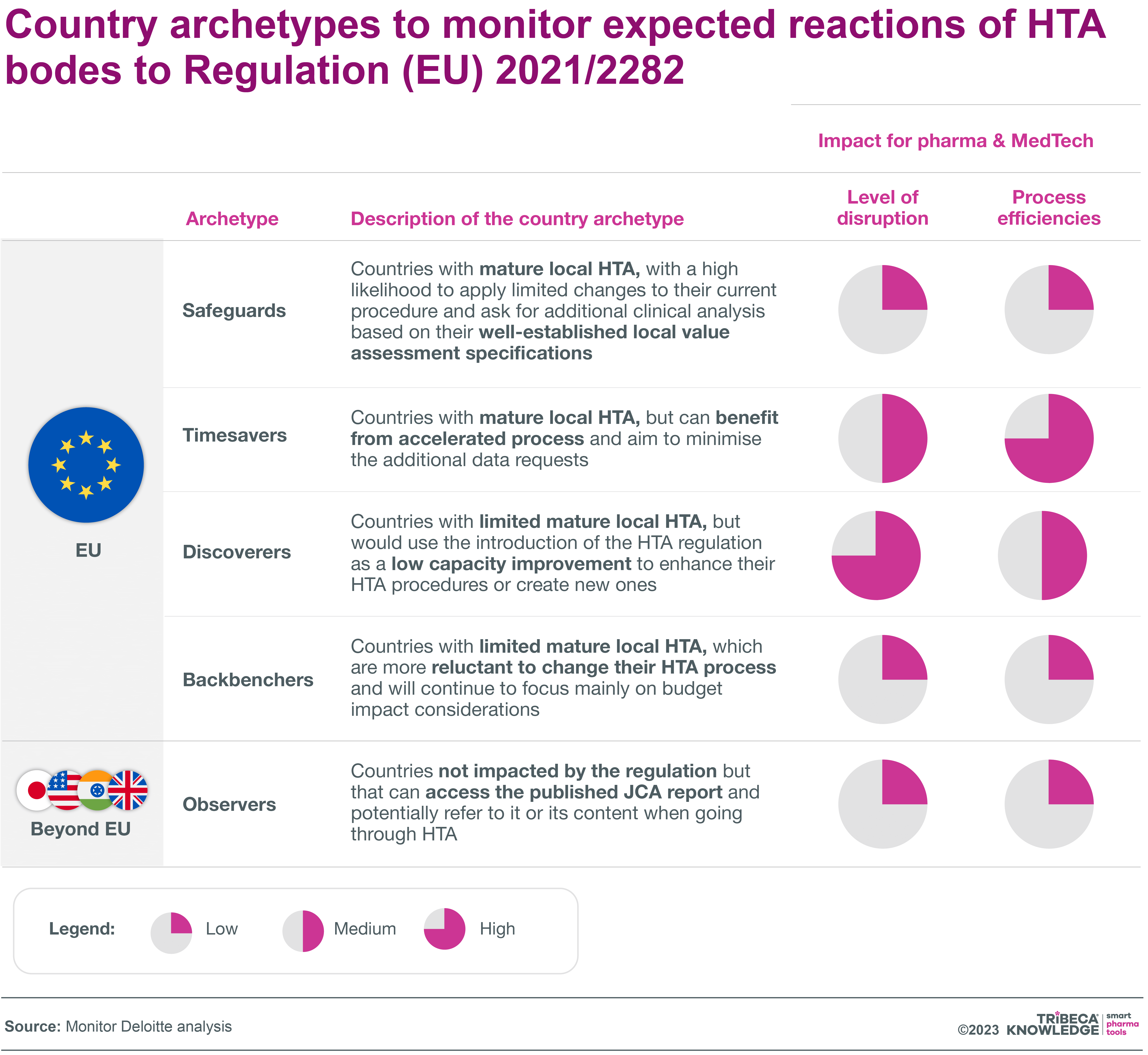
Fuente: Análisis de Monitor Deloitte
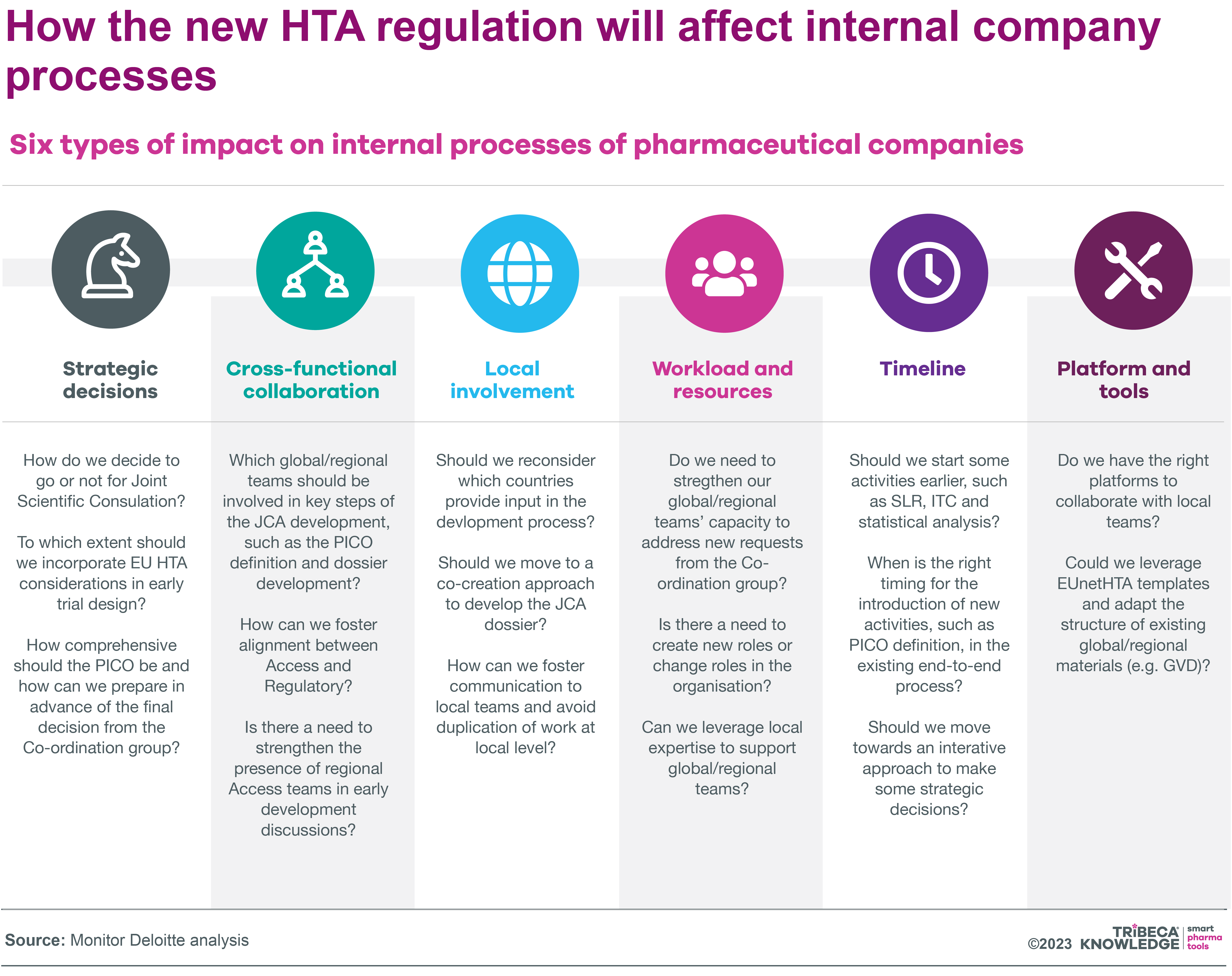
El informe de Deloitte también analiza cómo es probable que la regulación HTA afecte los procesos MA y HTA dentro de las empresas, identificando seis áreas clave de impacto:
- Toma de decisiones estratégicas en momentos críticos del desarrollo de fármacos : por ejemplo, decisiones de aprobación o no para consultas científicas conjuntas, equilibrio entre los requisitos de ETS y los requisitos reglamentarios para determinar el diseño temprano de ensayos clínicos.
- Trabajo multifuncional: g. Fortalecer la colaboración con equipos multifuncionales globales y regionales, y entre especialistas en acceso al mercado y regulatorios, desde el desarrollo inicial hasta la preparación y presentación de JCA.
- Participación local: g. construir un proceso centralizado en el que las necesidades locales se consideren desde el principio al diseñar un enfoque integral de eficacia clínica para los JCA y los expedientes locales de ETS.
- Carga de trabajo y recursos : por ejemplo, reevaluar los compromisos de tiempo y carga de trabajo de los equipos globales y regionales, desarrollar capacidades de previsión de capacidades, crear nuevos roles para facilitar y coordinar la preparación y presentación de la JCA.
- Plazos: g. acelerar las actividades previas al lanzamiento existentes para dar cabida a la preparación y presentación de la JCA en paralelo con el proceso de aprobación regulatoria; alinearlos con nuevas actividades como la colaboración con expertos clínicos de la UE.
- Plataformas y herramientas : por ejemplo, repensar la tecnología existente y las plataformas de colaboración para avanzar en la presentación y entrega de JCA, redefinir las estructuras de documentos globales basadas en plantillas EUnetHTA.
Fuente: Análisis de Monitor Deloitte
PICO = Población, Intervención, Comparación, Resultado (formato estándar para definir preguntas de investigación)
Como enfatiza Deloitte, ahora es el momento de actuar sobre estas cuestiones, incluso si persisten incertidumbres en torno a las metodologías finales, los documentos de orientación o los cronogramas para los procesos centralizados de ETS. Las empresas que deseen optimizar sus operaciones de HTA y aumentar la velocidad de comercialización en Europa a partir de 2025 deberían tomar en serio estas recomendaciones.
El compromiso corporativo oportuno con nuevos canales para la evaluación de tecnologías sanitarias en toda la UE, tanto externa como internamente, es vital si las empresas quieren protegerse contra evaluaciones del mínimo común denominador o procesos mal alineados. De lo contrario, la regulación de la UE corre el riesgo de agregar complejidad sin racionalizar los plazos, mejorar los resultados o lograr una armonización de las ETS realmente significativa y remunerativa en todos los estados miembros.
Lo que nos espera
Este blog sobre tendencias farmacéuticas para 2024 ha esbozado algunas consideraciones importantes para los gerentes de acceso al mercado y sus equipos mientras se preparan para otro año de cambios y consolidación en el mercado. Como siempre, el diablo está en los detalles, y los intereses farmacéuticos están trabajando arduamente para intentar limitar el daño potencial a sus negocios y a la causa más amplia de la innovación farmacológica abundante, disponible y asequible.
No obstante, está claro que, incluso con los sistemas de salud recuperándose de las circunstancias extraordinarias de la COVID-19, las incertidumbres en el momento, la coherencia, los criterios y los resultados de la ETS, los precios y los procedimientos de reembolso seguirán moldeando el acceso al mercado y las estrategias de lanzamiento de nuevos medicamentos. en el futuro previsible. Esto exige una transformación digital avanzada, estrategias de acceso al mercado mejor informadas y con mayor capacidad de respuesta, y una ejecución de lanzamiento mejor alineada pero ágil.
El software de preparación para el lanzamiento, como SmartLaunch™ , o las herramientas digitales orientadas al acceso al mercado, como SmartAccess™ , pueden ayudar a gestionar los cronogramas cambiantes de manera más eficiente, al tiempo que brindan visibilidad en tiempo real de la HTA, el reembolso y el estado del lanzamiento en múltiples países y niveles corporativos. Dados los desafíos apremiantes destacados en este análisis de la industria farmacéutica para 2024, las empresas deben tener aún más confianza en que sus productos pueden lanzarse y adoptarse en el mercado con el máximo impacto posible.
Una forma de hacerlo es optimizando las soluciones digitales, y específicamente el software de colaboración, para mantener la alineación en todos los ámbitos en el lanzamiento de productos farmacéuticos y la planificación y ejecución del acceso al mercado . Una visión de 360° y en tiempo real del progreso y los desafíos de un mercado a otro ayudará a garantizar que 2024 sea un año en el que los obstáculos al acceso al mercado no sólo se asimilen y comprendan plenamente, sino que también se aprovechen para ofrecer ventajas competitivas a las empresas y resultados verdaderamente beneficiosos para las empresas. sistemas de salud y pacientes.
Por: André Moa / Tribeca Knowledge: https://www.tribecaknowledge.com/